The National Institute of Standards and Technology added 41 new cannabinoid spectra to its mass-spectral library (Standard Reference Database 1A), increasing cannabis-related entries to 121. The update emphasizes rare side-chain homologs and derivatives to improve identification of uncommon cannabis compounds for forensics, biomedical research, food science and environmental testing.
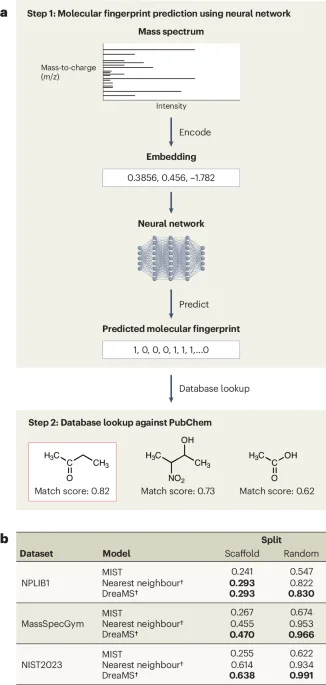
ML models for small-molecule structure elucidation from LC–MS/MS perform poorly compared with simple baselines due to generalization gaps across experimental conditions, ignored peak intensities, and unseen fragment formulas. Scaffold-split evaluations show nearest-neighbor retrieval often outperforms top models like MIST and DreaMS, revealing weak real-world generalization. Data-attribution analyses indicate the problems arise from both data and model design, prompting calls for domain-aware architectures, standardized datasets, and benchmarks that move beyond fingerprint-based, NLP-inspired translation toward chemistry-informed approaches.
Rockefeller University researchers unveiled MultiQ-IT, a parallel ion-trap mass spectrometer prototype that routes ions through hundreds of openings to analyze billions of molecules at once, boosting sensitivity and throughput by up to two orders of magnitude and enabling deeper single-cell proteomics and metabolomics—though it remains a proof-of-concept rather than a commercial instrument.
Testing two ferrous-looking pieces from Spain's Treasure of Villena (c. 1400–1200 BCE) indicates they were made from meteoritic iron, inferred from elevated nickel content measured by mass spectrometry; this places meteoritic iron among Iberian Bronze Age metalwork and suggests ironworking began earlier in Iberia than previously thought, though corrosion limits conclusive proof and further non-invasive analyses are planned.
Researchers at Stellenbosch University analyzed three cannabis strains and found 79 phenolic compounds, 25 of which were new to Cannabis, including 16 tentatively classified as flavoalkaloids—the first evidence of this rare group in Cannabis leaves. Using two-dimensional liquid chromatography and high-resolution mass spectrometry, they observed that these flavoalkaloids are concentrated mainly in leaves of one strain, underscoring substantial chemical variation and suggesting that cannabis leafy material—often discarded—may have unexplored medical potential beyond cannabinoids.
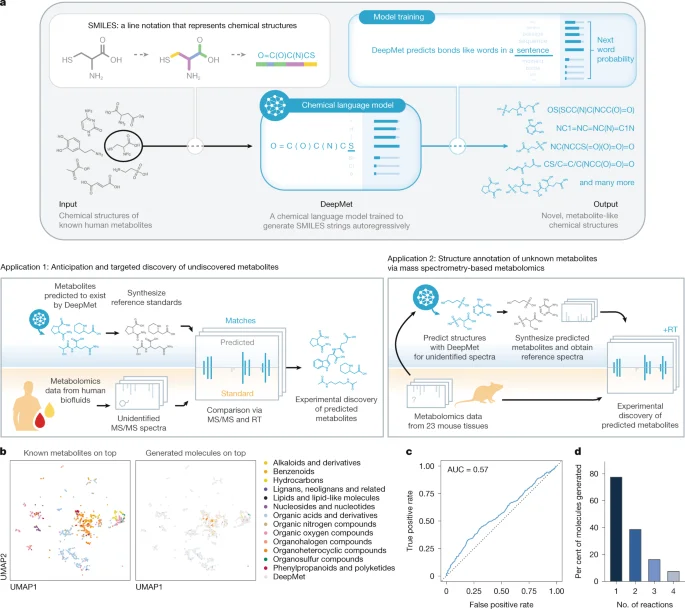
A new chemical language-model approach, DeepMet, learns from known human metabolites to generate metabolite-like structures and prioritize plausible, yet-unrecognized mammalian metabolites. By coupling DeepMet with mass-spec data and MS/MS prediction (CFM-ID), the method enables de novo generation and targeted discovery of metabolites, identifying 16 previously unrecognized mouse tissue metabolites and 17 metabolites in human biofluids, and correctly predicting 252 of 313 HMDB 5.0 additions (81%). The team further improves annotation with a meta-learning framework that integrates retention times and isotope patterns, achieving about 70% accuracy in a mouse dataset. They also release a web app and Snakemake pipeline to extend the approach, highlighting DeepMet’s potential to fill gaps in mammalian metabolome maps while noting limitations such as its focus on metabolite-like chemical space and isomer ambiguity.
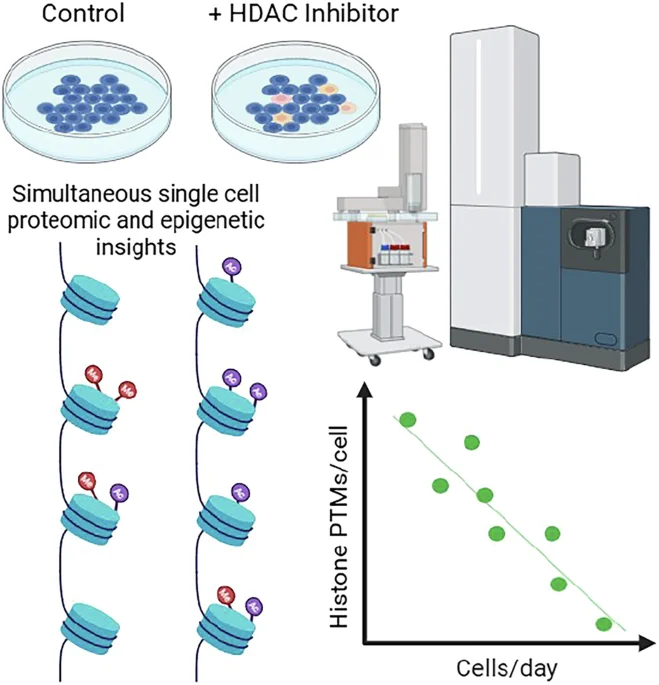
The article discusses advanced methods for analyzing histone deacetylase inhibition effects in human cells through simultaneous single-cell proteomics and epigenetic profiling, utilizing cutting-edge mass spectrometry and sequencing technologies.
Scientists at Yale have for the first time precisely measured the time it takes for protons to move through a small chain of water molecules, using a specialized mass spectrometer, providing new benchmarks for understanding proton transfer in chemistry.
Researchers from Oxford have developed a novel AEC-MS method for large-scale analysis of polar and ionic metabolites in biological samples, enhancing capabilities in metabolomics research and enabling new applications in health and disease studies.
Scientists have developed a groundbreaking method to extract and analyze proteins from preserved soft tissues, including human brains up to 300 years old, using urea and mass spectrometry, opening new avenues for understanding evolution, diet, and physiology from ancient biological archives.
Chemists have demonstrated the use of mass spectrometry to separate chiral molecules, which exist as mirror-image structures with different properties. This breakthrough could streamline the laborious process of separating enantiomers, crucial in drug discovery, by allowing for quick determination of enantiomeric excess and confirmation of molecular structures. The technique, described in Science, has the potential to simplify and expedite the preparation of pure samples of enantiomers in larger quantities, with implications for drug design and discovery.
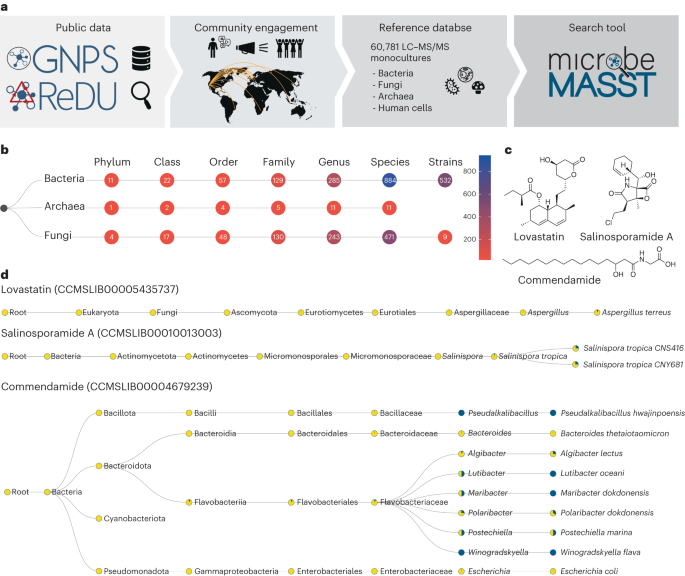
microbeMASST is a new search tool that leverages public mass spectrometry repository data to identify the microbial origin of known and unknown metabolites and map them to their microbial producers. It contains a reference database of microbial monocultures and allows users to search tandem mass spectrometry (MS/MS) spectra obtained from their experiments against the repository. The tool provides search results within seconds and displays them in interactive taxonomic trees, enabling users to filter results and access additional information. It has been used to identify microbial metabolites in mouse datasets and has the potential to enhance understanding of microbial metabolites across various ecosystems.
Researchers from the Skaggs School of Pharmacy and Pharmaceutical Sciences at the University of California San Diego have developed a technique called reverse metabolomics to identify chemical structures in the human body. By analyzing mass spectrometry data, the researchers were able to map the metabolic pathways of various compounds, including those produced by the human microbiome. This approach has the potential to advance our understanding of human health and disease, as well as aid in the discovery of new therapeutic targets.
Soft-landing mass spectrometry, a technique that gently lands intact proteins for analysis, shows promise in simplifying protein structure determination for cryo-electron microscopy (cryo-EM). By minimizing damage to proteins during the landing process, researchers have achieved near-atomic-resolution cryo-EM structures for proteins. This method could revolutionize protein sample preparation, allowing for the generation of high-resolution protein structures with greater precision and efficiency. However, further research is needed to optimize the technique and ensure that proteins retain their natural structure throughout the process. Soft-landing mass spectrometry also holds potential for single-molecule protein analysis and other structural analysis methods.
Scientists at the Department of Energy's Pacific Northwest National Laboratory are developing new mass spectrometry techniques to identify the 99% of chemical compounds that have not yet been characterized. By combining two high-resolution instruments, they aim to unlock potential cures for diseases, tackle climate change, and identify new chemical threats. This research is part of the m/q Initiative, which seeks to explore the vast sea of unknown compounds and revolutionize the field of mass spectrometry. The new technique allows for faster and more accurate measurements of chemical compounds, providing valuable information about their structure and potential applications.