Weizmann Institute researchers showed that deleting MTCH2 (Mitch) in human cells triggers a hypermetabolic state: mitochondria burn more fuel, especially fats, while fat storage and fat-cell formation are reduced, shedding light on MTCH2 as a regulator of cellular energy and obesity—though translating this into safe human therapies will require extensive follow-up.
Researchers identify MTCH2 ('Mitch') as a regulator of mitochondrial fusion; deleting Mitch in human cells fragments mitochondria and increases energy demand, boosting fat and carbohydrate breakdown and reducing new fat-cell formation. In mice, Mitch suppression in muscle improves endurance and protects against obesity. The findings point to Mitch as a potential target to enhance fat burning while limiting fat storage, though the work is early-stage and preclinical.
Healthy but sedentary adults show early cellular energy declines: mitochondrial efficiency drops 28–36%, MPC1 protein is 49% lower, and CPT1 activity is halved, alongside 38% lower VO2 max and 60% higher lactate during exertion, indicating a pre-disease shift in fuel processing that regular exercise may help prevent by maintaining mitochondrial fuel-switching (metabolic flexibility).
Aging appears to slow mitochondria partly because the lipid phosphatidylcholine declines; dietary supplementation restored mitochondrial function in worm models, and in human tissue higher phosphatidylcholine levels aligned with healthier aging, with notable sex differences around menopause. The study suggests boosting this lipid could counter mitochondrial aging and warrants further molecular study; published in Nature Communications.
A comparative genetic study of the two-toed sloth suggests that jumping genes and mitochondrial/metabolic gene networks form backup systems that help sustain the animal’s ultra-low-energy lifestyle, shedding light on how cells manage energy and potentially informing human health, aging, and tissue preservation.
Vitamin B12 is needed in trace amounts but deficiency is common, especially among older adults. Aging can reduce stomach acid and intrinsic factor, impairing B12 absorption from animal foods and causing fatigue, numbness, balance or memory problems. Testing and guidance are advised for those at risk; injections treat confirmed deficiency but don’t boost energy in people with normal B12. Recent research links B12 to mitochondrial energy in muscle and aging, but it does not reverse aging.
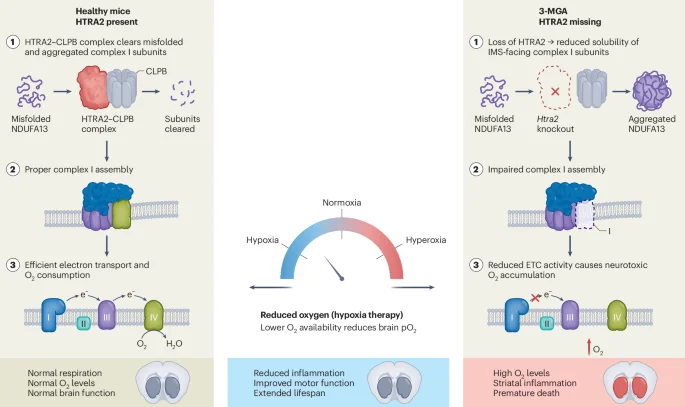
Scientists identify the enzyme GRK2, and its inactive form, as a new target in Alzheimer's disease. In mouse models and some human brain tissue, inactive GRK2 accumulates with mitochondria, boosting amyloid-beta production and harming mitochondrial function. The compound dubbed Compound 10 prevents GRK2 from clumping, restoring mitochondrial health, reducing amyloid-beta, and slowing dementia progression in mice, marking a potential new therapeutic avenue that still needs validation in humans.
Two micrograms of vitamin B12 are enough daily, but deficiency is common—especially in older adults with reduced stomach acid or absorption issues. B12 is essential for DNA and mitochondrial energy in cells, and newer research links low B12 to impaired mitochondrial energy production in muscle, which may help explain fatigue even before anemia appears. While B12 injections treat diagnosed deficiency, they don’t boost energy in people with normal B12 levels, so persistent tiredness should prompt investigation of underlying causes and absorption issues.
New research from the Leibniz Institute on Aging links age-related mitochondrial decline to reduced membrane phosphatidylcholine, which makes mitochondrial networks more fragmented and less adaptable. In Caenorhabditis elegans, impairing phosphatidylcholine production induced aging-like mitochondrial changes, while providing phosphatidylcholine or its precursor choline restored youthful mitochondrial structure and function within two days. The study integrated worm experiments, human cell data, and aging datasets, suggesting that lipid changes—not just genetic damage—drive mitochondrial aging and that dietary or metabolic interventions could modestly slow aging, though translation to humans requires further work.
ETH Zurich researchers identify inactive GRK2 aggregates that disrupt mitochondrial energy and drive amyloid beta buildup in Alzheimer’s, forming a damaging feedback loop. They developed Compound 10, which blocks GRK2 aggregation, preserves mitochondrial function, reduces amyloid beta, and slows nerve-cell loss in mice, suggesting a new upstream therapeutic approach that could complement existing treatments; a patent has been filed and industry partnerships are being pursued.
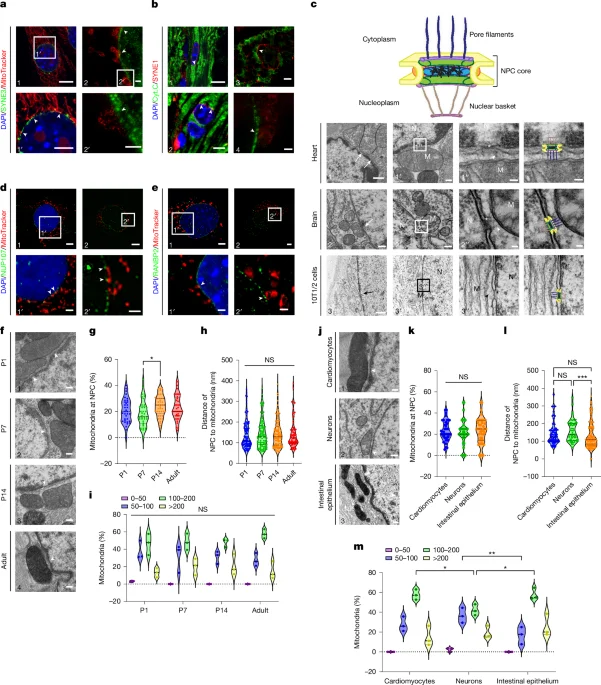
Scientists identify direct mitochondria–nuclear pore contacts mediated by VDAC1 and the RANBP2 C-terminal domain. Disrupting this interaction reduces mitochondria–nucleus proximity, lowers nuclear ATP and phosphocreatine, and alters the nuclear phosphoproteome, shifting pathways linked to histone modification, differentiation, and transcription. In vivo truncation of RANBP2’s CTD causes embryonic cardiac and neural crest defects, underscoring a vital mitochondria–nucleus communication axis that regulates nuclear energetics and cellular differentiation.
Almost 93, Emma Mazzenga’s elite sprinting performance is accompanied by aerobic capacity and neuromuscular health that resemble those of active younger adults, as researchers compare her metrics to younger athletes to understand what aspects of aging can be slowed by training. While she shows age-related leg mass loss and some fast-twitch fiber atrophy, her mitochondria function well and nerve connections are largely preserved. Her regimen—brief, high-intensity sprint workouts three times a week plus daily walking and light resistance—offers clues about the potential for exercise to slow aging, though many questions remain about training type, genetics, and diet as scientists repeat tests to track changes from age 91 to 93.
ETH Zurich researchers identified that inactivated GRK2 aggregates in dementia brains damage mitochondrial function and accelerate amyloid-beta production, creating a self-perpetuating cycle. They developed Compound 10 to prevent GRK2 aggregation, which improved mitochondrial energy, reduced amyloid-beta, and extended survival in Alzheimer’s mouse models, with additional systemic anti-aging benefits observed. The basic research is complete and a patent has been filed; industry partnership is sought to advance toward clinical development.
Texas A&M researchers, backed by a $2.9 million NIH grant, are investigating how a father's alcohol consumption before conception can imprint non-genetic signals in sperm that disrupt offspring mitochondrial function, potentially leading to birth defects, chronic disease, and accelerated aging, while exploring interactions with maternal exposure and implications for other environmental stressors.